I ran across an interesting Agile v. FDA discussion the other day. For those that are not familiar with what a FDA regulated product means, I'll give a brief overview.
In order to market and sell a medical device in this country -- OUS (outside US) medical device regulations are different -- you must have FDA approval. Most of the time, this involves a FDA Premarket Notification 510(k) submission. Your company must be registered with the FDA and is subject to periodic on-site visits by FDA inspectors to audit your quality system records. There are two important points here:
- Getting approval: In order to sell your device you not only have to prove safety and effectiveness, but you also have to demonstrate that the design and development of the device -- including the software -- follow Quality System Regulations. The Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices details these requirements.
- Keeping approval: After you receive 510(k) approval the FDA can pull your device off the market (the dreaded "recall") at any time due to complaints or unsatisfactory audit results.
What this means is that your on-going software development process must adhere to a well defined quality system process. As noted in the discussion, the FDA guidance does not dictate that a particular process must be used. The quality system process itself is designed by you -- actually, your entire company -- and simply needs to ensure that you are designing and building a quality product. The difficult part is that you have to be able to prove to the outside world that you have actually followed that process.
I've spent just about my entire career developing software for devices that were under FDA regulatory control. In the old days (pre ~1996) the FDA did not have a good concept of the software development process as part of it's quality system regulations and inspectors did not usually scrutinize software design and development. Nowadays, the FDA has a much clearer understanding of the software development process. It's that understanding that is the one of the central issues with respect to adopting an Agile development process for software in medical devices.
Let's first look at the FDA Good Manufacturing Practice (GMP - Quality System Regulation) requirements. It's Subpart C--Design Control (§ 820.30) that's of primary interest. Here's the outline:
- General
- Design and development planning
- Design input (specifications)
- Design output (coding)
- Design review
- Design verification (Was the product built right?)
- Design validation (Was the right product built?)
- Design transfer
- Design changes
- Design history file
The critical issue for the software development process is that each of the items 3-7 require a formal review and approval step. This is the reason why most companies that develop medical device software have chosen to use a quality system process that follows the Waterfall model for their development.
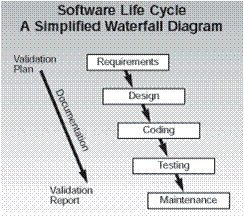
This sequential approach is a natural fit for allowing you to review and document each step in the process.
Now let's look at the Agile process. One of the better descriptions of the Agile development process is 'The New Methodology' by Martin Fowler. There a number of flavors of Agile (SCRUM, Extreme Programming (XP), etc.) that all try to encompass the Agile Manifesto. One advantage that the Agile process has over the Waterfall approach is it's ability to adapt to the unpredictability of requirements and changing customer needs. This is handled through the use of Iterations. From the Martin Fowler paper:
The key to iterative development is to frequently produce working versions of the final system that have a subset of the required features. These working systems are short on functionality, but should otherwise be faithful to the demands of the final system. They should be fully integrated and as carefully tested as a final delivery.
The purpose of the iterative development is to facilitate requirements changes between each iteration. Agile Methodologies in a Validated Setting by Frank Jacquette proposes some steps to accomplish the use of iterative development in a FDA regulated environment.
Let's assume that an Agile development process would be able to produce higher quality medical device software and that because of the customer focus of this process the resultant product would better meet market needs. Even with these assumptions, I think there are two major issues that need to be addressed:
- I just don't see how a cost-effective quality system process implementation can be accomplished. Even if (and it's a big if) the actual overall software development time was shorter, the extra costs incurred by the additional process controls, documentation and testing required for each iteration would far exceed those savings.
- The last point Mr. Jacquette makes is the other issue:
Take the time to explain agile methodologies to your regulatory specialists and work hard to gain their understanding and agreement, because the burden of proof will be on them and you if an FDA auditor decides to take a peek under the hood.
This seems like a huge risk to me.
The first question becomes: Is the possibility of an improved product (features and quality) worth the additional cost? I suppose if you had a development team with extensive Agile experience you could make the argument that it would be worth it. If not, I think the ROI (return on investment) analysis would be a difficult one to make.
The second question is a big unknown, which is why I think the risk is high. My experience with FDA auditors is generally good. They are professionals that are focused on getting a very specific job done. Since their interest is the entire quality system, the typical audit is a whirlwind affair as it is. The amount of time spent on design control (auditing the Design History File) is usually minimal. Even if you had received 510(k) approval with an Agile design control process, having to take the time to explain to an on-site FDA auditor (who in all likelihood has never seen your 510(k)) a methodology they probably have never even heard of is reason to worry.
Conclusion:
It seems to me that Agile methodologies have a long way to go before we see them commonly used in medical device software development. I've searched around and have found nothing to make me think that there is even a trend in this direction. Maybe it's that Agile processes are just too new. They seem popular as a presentation topic (I've been to several), but I wonder how prevalent Agile is even in mainstream software development?
If you are (or have ever been) part of an Agile development team for a FDA regulated product I'd love to here about your experiences and how you were able to resolve the types of issues presented here.
Thanks!
